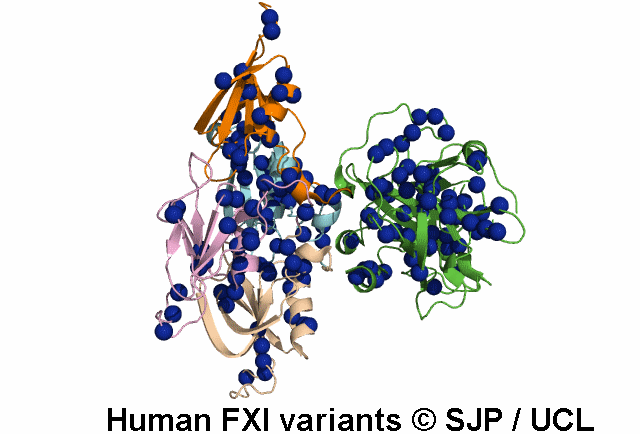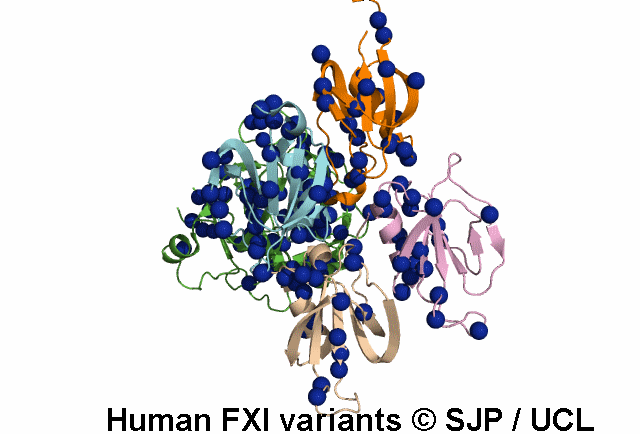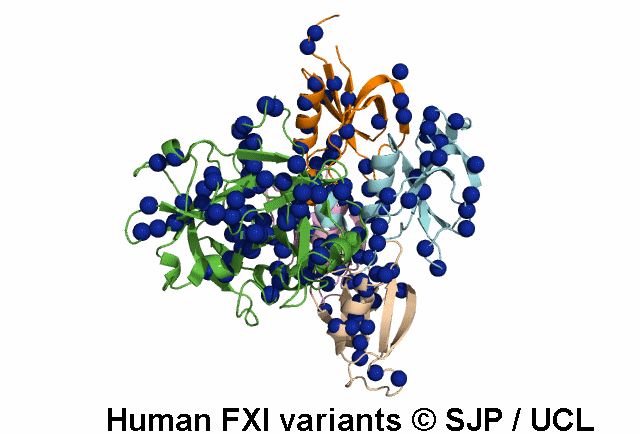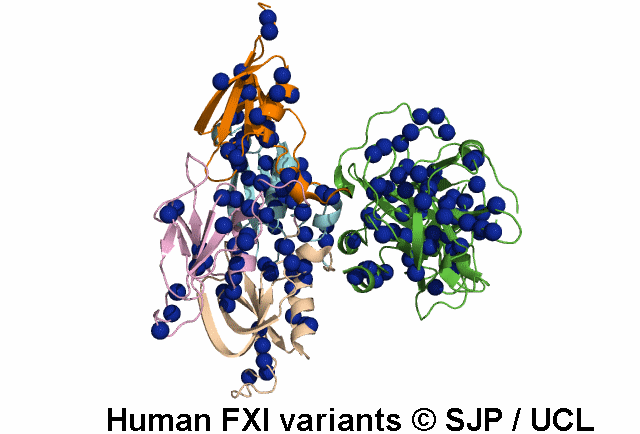
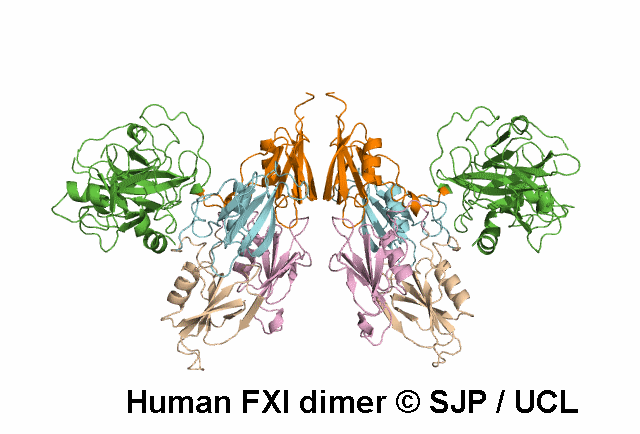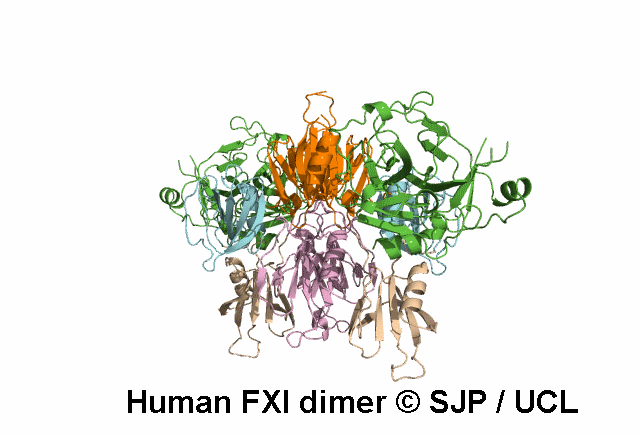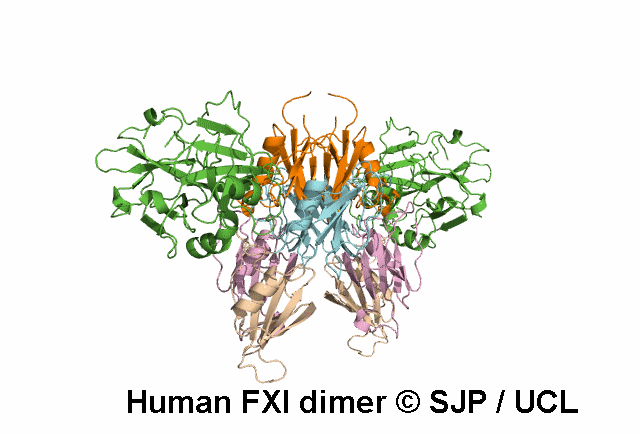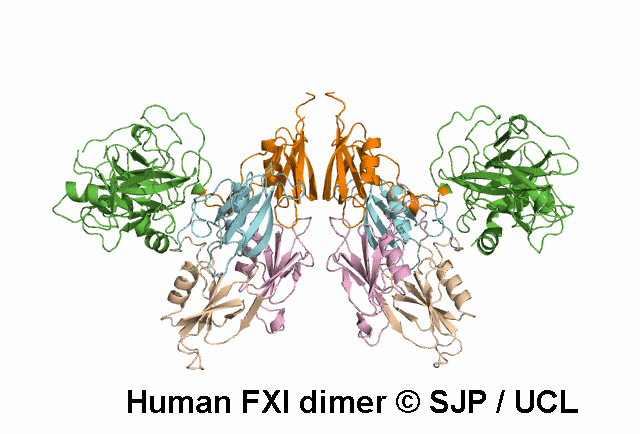FXI deficiency is a rare bleeding disorder, occurring at a frequency of one in a million in the general population.
The condition is more common among defined populations such as French Basques and Jewish communities, with Ashkenazi Jewish populations having the highest
prevalence of approximately 10%.
FXI deficiency in Jewish populations has been well-studied with four causative mutations identified. These mutations are referred to as Jewish mutations
Types I-IV. This nomenclature should not be confused with phenotypic classification of Type I (low activity and low antigen) and Type II (low activity and normal antigen).
All four Jewish mutations are in fact Type I.
- Jewish mutations Type II (Glu135Stop - HGVS) and Type III (Phe301Leu - HGVS) are the two most common variants.
- Jewish Type I (a G to A transition at the donor splice site in intron N) and Type IV (a 14bp deletion at the exon 14 / intron N splice site) both affect splicing in the
catalytic SP domain and are seen less frequently.
DISTRIBUTION OF VARIANTS
An increasing number of variants are being reported in non-Jewish patients. Most of these are missense
point mutations and occur throughout the F11 gene clustering in the Ap4 and SP domains.
VARIANT DATA
The variants associated with FXI deficiency have been reported with varying amounts of data.
- Coagulation Activity
The coagulation activity of FXI (FXI:C) is usually measured with a one-stage clotting assay.
- Antigen Level
The level of FXI protein in plasm (FXI:Ag) is usually assayed by ELISA with a FXI polyclonal antibody.
- Expression Data
A number of variants have been published with expression studies, comparing FXI Antigen and mRNA levels in cell medium and cell lysates.